Проявления на коже при рандю ослера. Можно ли успешно бороться с болезнью Рандю-Ослера? Смотреть что такое "Ослера - Рандю болезнь" в других словарях
В статье приведены основные данные о болезни Рандю-Ослера. Когда и как проявляется эта патология, особенности разных форм локализации патологических образований сосудов. Чем опасен этот синдром, и какие методы лечения используют. Можно ли полностью излечиться от заболевания.
Дата публикации статьи: 01.07.2017
Дата обновления статьи: 29.05.2019
Болезнью Рандю-Ослера называют заболевание, которое приводит к нарушению строения сосудистой стенки в процессе развития эмбриона. Проявляется множественными сосудистыми новообразованиями на коже, слизистых и серозных оболочках (телеангиэктазии или ангиомы), формированием патологических связей между артериальной и венозной системой во внутренних органах (мальформации).
Проявление телеангиэктазии на лице
Заболевание носит наследственный характер, то есть связано с нарушениями в строении генов и передается ребенку от родителей. Редко встречаются спонтанные или спорадические случаи болезни, возникшие на фоне мутации (патологического изменения) генов в процессе развития эмбриона.
По-другому синдром называют:
- Наследственная или семейная геморрагическая телеангиэктазия.
- Генерализованный ангиоматоз.
- Геморрагический семейный ангиоматоз.
Диагностируют у одного человека на 50 тыс. населения. Несмотря на генетический характер заболевания, оно передается не во всех случаях и может протекать с разной степенью тяжести даже в пределах одной семьи.
При наследственной телеангиэктазии происходит нарушение строения стенки мелких капилляров в виде недоразвития двух из трех оболочек. Патологический очаг представлен только внутренней выстилкой, или интимой, мышечный и серозный слой полностью отсутствуют. Такой истонченный участок стенки под действием давления крови выбухает, формируя ангиому.
Интима, которой представлена стенка образования, – очень хрупкая структура, она легко травмируется, что ведет к кровотечению. Отсутствие в зоне травматизации сосуда основных оболочек не дает ему сократиться и «склеить» дефект. В результате развивается длительное и обильное кровотечение, приводящее к снижению количества гемоглобина в крови заболевшего (анемии).
При развитии таких дефектов в сосудах внутренних органов (печень, головной мозг, легкие) между участками с истонченной стенкой формируются патологические связи, или шунты, по которым происходит сброс венозной крови в артериальную или наоборот. Это приводит к недостаточному кровоснабжению участков органа и нарушает его функцию.
Генерализованный ангиоматоз – опасное заболевание, которое может привести к гибели заболевшего в результате:
- обильного кровотечения;
- острого нарушения кровотока в головном мозге;
- дыхательной или сердечной недостаточности.
Учитывая, что причиной патологии является генетический дефект, излечиться от заболевания невозможно. Процесс формирования патологических деформаций сосудов продолжается в течение всей жизни. Пациенты регулярно нуждаются в медицинской помощи, требуют ограничительных мероприятий в обычной жизни и на работе. Тяжесть проявлений и продолжительность жизни зависят от объема поражения сосудистого русла, особенно внутренних органов.
Наблюдают и лечат пациентов с такой патологией врачи многих специализаций, что связано с обширностью поражения организма. Заболевшие часто бывают у гематологов, сосудистых и общих хирургов, отоларингологов, терапевтов. Могут требовать помощи торакальных и нейрохирургов.
Классификация
В зависимости от стадии деформации стенки сосуда выделяют основные типы телеангиэктазий.
| Тип | Описание |
|---|---|
| Ранний | Изменения проявляются в виде мелких, разной формы пятен розоватого цвета
|
| Промежуточный | Пятна превращаются в образования с множественными извитыми лучами, похожими на лапки паука
|
| Поздний | На месте плоского паукообразного образования развивается плотный узел до 0,5–0,7 см яркого красного цвета, возвышающийся над поверхностью на 1–3 см.
|



Симптомы болезни
Несмотря на генетическую основу, заболевание обычно проявляет себя в промежутке между шестью и десятью годами жизни ребенка, когда возникают первые сосудистые звездочки на коже и слизистых. Синдром активно прогрессирует в период полового созревания с 12–14 до 18–20 лет. К этому возрасту у пациентов на коже и слизистых можно увидеть все три типа телеангиэктазий.
Патологическое изменение сосудов продолжается на протяжении всей жизни. И если в 16 лет проявления болезни есть у 71 % пациентов, то к 40 годам более 90 % заболевших регулярно обращаются за помощью.
Симптоматика зависит от интенсивности поражения сосудистой системы. Всех пациентов беспокоят кровотечения из сосудистых образований, но их продолжительность и тяжесть различны: от незначительного дискомфорта до крайней степени анемии, требующей длительного стационарного лечения. Независимо от этого, ограничительный режим рекомендован всем заболевшим.
Особенность болезни Рандю-Ослера – обширность поражения сосудистой системы всего организма. Встречаемость изменений по системам и органам представлена в таблице.
Кожа и слизистые оболочки
Первыми проявлениями болезни всегда являются телеангиэктазы на слизистых оболочках. Частота поражения органов по ее уменьшению:
- ротовая полость;
- внутренняя поверхность век;
- желудок и кишечник.
Основная жалоба пациентов: повторные кровотечения, в 80–90 % носовые. Частота эпизодов может колебаться от одного раза в сутки до одного в месяц. Интенсивность кровотечения также носит индивидуальный характер: умеренное, с которым возможно справиться в домашних условиях, или тяжелое, требующее обращения в больницу.
Кровотечение из ангиом на веке проявляется кровавыми слезами.
Кожные телеангиэктазы локализуются преимущественно на лице (губах, крыльях носа), значительно реже – на теле и под ногтями. Их отличие от других сосудистых образований (петехий): при надавливании бледнеют, а после прекращения давления быстро восстанавливают свой цвет.
Кожные изменения присоединяются в течение года после первого эпизода кровотечения.
Желудочно-кишечный тракт и печень
Проявления поражения желудочно-кишечного тракта есть у 10–40 % заболевших. Они возникают в более поздние сроки, чем носовые кровотечения, и длительно протекают без каких-либо симптомов.
Легкие
Большая часть пациентов с небольшим шунтированием крови в сосудистой системе легких никаких жалоб не предъявляет. При более обширном поражении проявления болезни включают:
- Нарастающее затруднение дыхания при физическом напряжении.
- Периодические приступы кашля с кровавой мокротой и синюшностью лица.
- Могут быть приступы удушья при смене положения тела (ухудшение – лежа);
У симптомных заболевших постепенно развивается .
Центральная нервная система
Патологические сосудистые шунты в головном мозге (мальформации) встречаются у 10 % пациентов с наследственной геморрагической телеангиэктазией, из них у 33 % поражение носит множественный характер.
Жалобы при таком расположении образований могут включать:
- мигренеподобные боли;
- периодические судороги;
- выпадение чувствительности или движений на отдельных участках тела;
- нарушения зрения, слуха, речи.
Диагностика
Болезнь Рандю-Ослера относится к заболеваниям, которые устанавливаются по данным клинических проявлений. Дополнительные методы исследования назначают в спорных случаях и с целью выявления поражения внутренних органов.
Классические критерии диагноза генерализованного ангиоматоза:
- множественные эпизоды носовых кровотечений;
- телеангиэктазии на коже и видимых слизистых;
- мальформации и ангиомы внутренних органов (даже без кровотечений);
- установленный диагноз у родственников первой линии (отец, мать, братья и сестры).
Совпадение по трем и более пунктам говорит о наличии болезни Рандю-Ослера. Если пациент соответствует только двум критериям – это подозрение на синдром. Один признак – болезни нет.
Дополнительные исследования при подозрении на семейный ангиоматоз (или если диагноз установлен только клинически) обязательно включают ряд процедур.
Эндоскопическое обследование
 Общий вид всех эндоскопических исследований
Общий вид всех эндоскопических исследований
Лучевые обследования
Лабораторная диагностика
Лечение
Заболевание связано с нарушениями в структуре генов, отвечающих за правильное развитие стенки сосудов, поэтому вылечиться от болезни Рандю-Ослера невозможно. Проводят только терапию возникших проявлений патологического процесса.
- Исключить применение противовоспалительных средств, таких как Аспирин, Кетопрофен, Нимесулид и пр.
- Не употреблять острую пищу и маринады.
- Исключить алкогольную продукцию в любой форме.
- Нормализовать режим труда и отдыха.
- Не работать в ночное время.
- Избегать стрессовых ситуаций.
- Не допускать физического перенапряжения.
- Контролировать уровень давления.
- Избегать любых травм носа и телеангиэктазов.
Остановка кровотечения
Терапевтические методы:
- тугая перевязка, тампонада носа: передняя и (или) задняя;
- местное применение лекарственных препаратов, которые останавливают кровотечение: Аминокапроновая кислота, Перекись водорода, Гемостатическая и Коллагеновая губки;
- внутривенное введение средств, улучшающих свертывание крови: Транексамовая кислота, Дицинон, Этамзилат, Аминокапроновая кислота;
- инфузии компонентов крови: свежезамороженная плазма, криопреципитат, факторы свертывания.
Хирургические методы:
- прижигание кровоточащего сосуда электричеством, лазером, жидким азотом;
- отслоение слизистой оболочки;
- перевязывание мелких и крупных сосудов, включая ветви сонной артерии;
- иссечение телеангиэктазов на коже и слизистых оболочках;
- открытые операции на органах для полного удаления внутренних мальформаций;
- малоинвазивные эндоваскулярные операции с эмболизацией (полным перекрытием просвета) сосудов в ангиомах.
 Проведение эндоваскулярной операции
Проведение эндоваскулярной операции
Коррекция нарушений после кровотечения
- Переливание компонентов крови для нормализации уровня гемоглобина.
- Введение железосодержащих лекарственных препаратов (внутримышечно или через рот в зависимости от тяжести анемии).
Прогноз
Заболевание у всех пациентов протекает в разной степени тяжести, что обусловлено степенью изменения генов, поэтому нет никаких точных цифр и сроков жизни для заболевших.
Излечения от наследственной геморрагической телеангиэктазии нет, процесс образования патологических ангиом на сосудах продолжается в течение всей жизни. Внутренние органы обычно поражаются после слизистых оболочек и кожного покрова. Опасными для жизни пациентов являются обильные кровотечения из мальформаций в печени и головном мозге. Редко встречаются фатальные эпизоды носовых и желудочно-кишечных кровотечений.
Все пациенты требуют ограничительного режима и пожизненного наблюдения с контролем показателей красной крови.
Болезнь Рандю-Ослера - это появление кровотечений из небольших капилляров (артериол или венул). Характерно, что данный процесс имеет не воспалительный характер. Чаще всего проявляется заболевание в виде сосудистых сеток (или звездочек). Но поражение сосудов возникает не только на поверхности кожного покрова. Также симптомы могут проявляться на слизистых оболочках других органов (бронхов, носа, рта, мочевого пузыря).
Болезнь Рандю-Ослера: клиника
Впервые данное заболевание описал выходец из Франции - Луи Мари Рандю. Полную же картину болезни предоставил английский специалист Уильям Ослер. Информацию дополнили и наблюдения Фредерика Вебера. Установлено, что данное заболевание передается по наследству. Ребенку достаточно унаследовать дефектный ген родителя. Страдают данным синдромом одинаково как мальчики, так и девочки. За развитие его отвечают следующие гены: первый отвечает за эндоглин, контролирует выработку гликопротеина. Второй относится к категории трансформирующих факторов роста. При их мутации сосуды теряют эластичность, наблюдаются дефекты их соединений, отклонения от нормы в строении эндотелиальных клеток. Стоит отметить, что болезнь Рандю-Ослера (диссеминированный гемангиоматоз) - состояние довольно редкое, а природа его происхождения не всегда понятна. Показатели распространенности болезни таковы: ею страдает 1 человек на 5000.
Первые симптомы развития болезни
Как правило, до 6 лет симптоматика заболевания не проявляется (несмотря на то что болезнь Рандю-Ослера передается по наследству). По достижении определенного возраста (6-10 лет) появляются первые признаки. На коже или слизистых других органов можно заметить появление ангиом (то есть опухолей сосудов). В результате дефектов капилляров и сосудов могут образовываться красные (или с оттенком синего) пятна, которые кровоточат. характеризуется и низкими показателями свертываемости крови. В результате этого возможны кровотечения (даже как следствие микротравмы). Порой единственным симптомом, который может указать на наличие заболевания, являются носовые кровотечения. Также у девочек могут быть обильные менструации. Из-за частых кровотечений развивается малокровие, уровень гемоглобина падает.
Стадии заболевания
Имеет несколько стадий. Для каждой из них характерны определенные симптомы. На ранней стадии поражения сосудов небольшие, имеют вид мелких пятен. Промежуточная степень характеризуется наличием сосудистых звездочек, «паучков». Узловатая форма - это появление красных (круглых или овальных) узелков, которые выступают над поверхностью на несколько миллиметров. Диаметр они имеют от 5 до 7 миллиметров. При надавливании данные образования становятся бледными (все типы). Потом же они наполняются кровью.

Классификация форм болезни
Как уже упоминалось, может носить наследственный характер болезнь Рандю-Ослера (фото проявлений недуга можно увидеть в этой статье), которая передается от родителей. Если таким синдромом страдают и папа, и мама, то у ребенка заболевание будет протекать в более тяжелой форме. Также существуют и спорадические случаи, которые возникают из-за мутации. В зависимости от места поражения специалисты выделяют назальный из носа), глоточный, кожный (могут кровоточить определенные участки кожи). Висцеральная форма характеризуется кровотечениями из внутренних органов. Также может наблюдаться и смешанный тип, при котором поражаются как кожные покровы, так и внутренние органы.

Болезнь Рандю-Ослера (шифр МКБ-10): при диагнозе
Прежде всего, главным фактором опасности является наследственность. Также болезнь могут спровоцировать медицинские препараты, которые принимает беременная женщина в первые три месяца. Небезопасны и инфекционные заболевания, перенесенные в данный период. Спровоцировать кровотечение могут следующие моменты: трение одежды о поврежденные участки, несбалансированное питание. Отсутствие в пище необходимых микроэлементов, которые способны укрепить сосуды, - вот одна из причин их слабости. Особенно это касается сторонников вегетарианства.

Методы диагностики
Формулировка клинического диагноза при болезни Рандю-Ослера базируется на осмотре физического состояния больного. Как правило, на лице, голове, слизистых можно увидеть телеангиэктазии. Это образования красного цвета, которые выступают над поверхностью кожного покрова. Располагаться такие дефекты могут и на внутренних органах. Необходим общий анализ крови, который может указать на наличие анемии. Биохимический анализ дает возможность распознать сопутствующие инфекции и болезни. Также производится анализ мочи. Если в ней будут обнаружены эритроциты, то можно говорить о том, что развивается кровотечение почек, мочевыводящих путей. Специалист проводит ряд тестов. Проба щипка дает возможность судить о подкожных кровоизлияниях. Для проведения теста кожу под ключицей сдавливают на некоторое время. Подобную информацию дает и проба жгута (он накладывается на область предплечья на несколько минут). Если существует подозрения на болезнь Рандю-Ослера, диагностика не обходится без проведения тестов на скорость свертываемости крови, длительность кровотечения (пробивается палец). Полости тела осматриваются с помощью эндоскопа. Если наружные симптомы отсутствуют, то проводится компьютерная томография. Данный метод позволяет увидеть любые нарушения во внутренних органах.

Лечение заболевания
Болезнь Рандю-Ослера лечение предусматривает следующее. Во-первых, это консервативная терапия. Суть ее заключается в применении ингибиторов для орошения поверхности, что кровоточит. Данные медикаменты препятствуют рассасыванию сгустка крови и останавливают таким образом кровотечение. Как показывает практика, другие лекарственные средства малоэффективны. Нередко требуется оперативное вмешательство. В ходе его проведения удаляется поврежденная часть сосуда. Ее может заменить протез (если это необходимо). При носовых кровотечениях неплохой результат дает криокоагуляция. Используя жидкий азот, замораживается участок сосуда. Менее эффективным является использование лазера для прижигания. В некоторых случаях необходима гормональная терапия.
(гемокомпонентная терапия)
В тех случаях, когда болезнь Рандю-Ослера вызывает большие кровопотери, необходимо использование донорской крови. Дефицит компонентов, отвечающих за свертывание, восполняет переливание плазмы (свежезамороженной). Такая терапия используется при состояниях, которые угрожают жизни человека. Также в подобных случаях может понадобиться переливание донорских тромбоцитов. В случаях если пациент имеет тяжелую форму анемии (очень низкий уровень гемоглобина) или наступила анемическая кома (утрата сознания из-за нехватки кислорода для головного мозга), проводится переливание эритроцитов из крови донора. Зачастую их освобождают от белков, находящихся на поверхности.

Возможные осложнения недуга
Болезнь Рандю-Ослера, патогенез ее развития и отсутствие эффективного лечения может привести к целому ряду негативных моментов. Во-первых, развивается анемия. Значительные кровопотери способны привести к анемической коме. Человек при этом находится без сознания и никак не реагирует на внешние раздражители. Возможно также и наступление паралича. Данное заболевание может привести к слепоте (при кровотечениях в области сетчатки). Состояние внутренних органов значительно ухудшается. Опасным побочным эффектом болезни может стать кровоизлияние в головной мозг. Однако вовремя выявленное заболевание и грамотная терапия позволяют спасти здоровье и жизнь пациенту.
Профилактические мероприятия
Первичные меры профилактики необходимы еще до обнаружения проблемы. Семьи, в которых у одного или нескольких членов диагностирована болезнь Рандю-Ослера, должны проводить консультации с генетиком. Бывают случаи, когда дается рекомендация воздержаться от Также стоит позаботиться о защитных силах организма. Хорошее действие окажут регулярные прогулки на улице, закаливание организма. Правильное питание со сбалансированным рационом также необходимо.

Вторичная профилактика заключается в регулярных осмотрах у специалистов. Ее целью является раннее выявление болезни. Если синдром уже диагностирован, то лечение должно быть своевременным и полноценным. При необходимости оперативного вмешательства пациент в обязательном порядке должен проконсультироваться с гематологом. Это позволит обезопасить человека от возникновения осложнений при операции (больших кровотечений). Каждый пациент и его близкие должны получить информацию о том, как оказывать доврачебную помощь при кровотечениях (как внутренних, так и наружных). Рекомендуется делать переливание донорских компонентов крови только в случаях
Существует патология, связанная с генетической предрасположенностью, которая несет за собой недостаточное развитие кровеносных сосудов, расположенных в различных местах и служит причиной геморрагического диатеза. Болезнь Рандю-Ослера-Вебера – это ангиомы и телеангиэктазии, которые распространяются на слизистой оболочке рта и носа, внутренних органах, кожном покрове, губах. Этот процесс сам по себе не воспалительный. У человека наблюдается самопроизвольные носовые и , причиной которых является анемия.
Врачи впервые узнали, что такое синдром Рандю-Ослера в конце 19 – начале 20-го столетия. Свое название недуг получил благодаря Ф.П. Веберу, У. Ослеру и А.Ж.Л.М. Рандю. К счастью, заболевание наблюдается нечасто, лишь 1/5000 людей, однако она не отличается по половому признаку и возрастных категорий.
Болезни по генетической предрасположенности вредны тем, что весьма сложно лечатся. Специалисты обеспечивают симптоматическое лечение, так как нет возможности воздействовать на измененный ген.
Повреждение идет сосудистой системы и характеризуется регулярными кровотечениями. Стенки капилляров становятся тонкими и слабыми, увеличивается просвет, формируются анастомозы артериально-венулярной системы.
Причины
Выяснить причину образования болезни Рандю-Ослера еще не удалось до конца. Как уже отмечалось ранее, основная причина – наследственный фактор. Мутирующие гены являются главной этиопатогенетической причиной болезни. Это явление приводит к возникновению нарушения эндотелия, а далее к его деформации, повреждению капилляров и кровотечений.
Около сосудов собираются гистиоциты и лейкоциты, дисфункция тромбоцитов, развивается фибринолиз. Очень быстро расширяются венулы и в конечном итоге пониженное кровяное давление и . Если слега повреждены аневризмы в артериях и венах, то происходят кровотечения. Повреждение сосудов передается по наследственности по аутосомно-доминантному фактору.
Существует ряд опасных факторов, которые приводят к образованию заболеванию у людей с генетической предрасположенностью: на первых порах беременности, инфекционные болезни у будущих мам, медикаменты. В результате таких факторов у малыша внутри утробы деформируется сосудистая структура.
Имеются различные причины болезни Рандю-Ослера, способные усилить кровотечения:
- Вредные привычки, в частности алкоголь;
- Депрессия и стрессы;
- Вегетарианство;
- Перенапряжение;
- Травмы механического характера;
- Нарушенный сон, работа в ночное время суток;
- Медикаментозная терапия (дезагрегантами – аспирин).
Усиление болезни схоже с гормональным сбоем у человека в момент полового созревания, беременности, с рождением ребенка.
Симптомы
Симптоматика заболевания обуславливается возникновением на кожном покрове . Болезнь Ослера-Рандю-Вебера очень легко различить сразу. Проступает телеангиэктазия (главный субстрат недуга). Первоначально формируется небольшое пятнышко. Оно шаг за шагом преобразовывается в узелок ярко-красного цвета, выступающий на коже и сильно отличающийся от здоровых участков кожи.

Самые частые места распространения у пациента – ротовая и носовая полости, ногти, голова, губы, слизистая оболочка трахеи, глотки, гортани, почек, желудка, бронхов, на пальцах рук, мочевыводящих путей.
Симптомы возникновения болезни Рандю-Ослера:
- Увеличивается вероятность желудочно-кишечных и ;
- Анемия, которая возникает в следствии усиленных кровотечений;
- Телеангиэктазия (пятна, которые кровоточат на теле больного или слизистых оболочках синюшного или красного оттенка);
- Часто кровоточащие ангиомы.
Больной часто страдает от внешних и , кровь прослеживается в моче, мокроте, кале. Носовое кровотечение может быть настолько сильным, что нередко возникает анемия, а порой человек даже умирает. Чтобы установить причину внутренних кровотечений, врачи используют эндоскопию.
Артериовенозные аневризмы, расположенные в легочных путях, сопровождаются одышкой, синим оттенком кожи, отхаркиванием кровью.
При сильных осложнениях болезнь Рандю-Ослера может стать причиной следующего:
- Потери зрения;
- Отека;
- ТЭЛА ();
- Инсультам и параличам;
- Миокардиодистрофии;
- Цирроза печени;
- Внутричерепного кровоизлияния;
- Анемической комы и .
Диагностика болезни Рандю-Ослера – это сперва исследование больного. Если обнаруживаются характерные телеангиэктазии, то легко определить, что это такое. Пробы щипка и жгута позволяют пронаблюдать кровоизлияния под самой кожей. Анализ крови предоставляет медикам возможность определить гипохромную анемию, мочи – микро или макрогематурия в результате нарушения сосудистой системы в мочевыводящих участках. В процессе коагулограммы выраженных изменений не наблюдается.
Диагностика сопровождается и иными способами обследования синдрома Рандю-Ослера:
- Рентген грудной клетки;
- УЗИ внутренних органов.

Лечение
Врачи назначают симптоматическое лечение болезни Рандю-Ослера. Это значит, что выписываются лекарственные препараты, направленные на устранение очага кровотечения. Пациент находится в стационарных условиях.
Традиционное лечение стремится остановить кровотечение:
- Пациенту назначаются препараты, которые не дают рассосаться , останавливают кровотечение.
- Источники кровотечения обрабатывают кровоостанавливающими препаратами, для внутреннего применения назначаются гемостатическое лечение – Этамзилит, Дицинон, Викасол, Аминокапроновая кислота, плюс средства, которые хорошо воздействуют на сосуды, регенирирующие и эпитализирующие препараты – витамины Е, С, Дексапантенол.
- Препараты, содерражие гемостатический эффект – Адроксон.
- Гормональное лечение нацелено на высокий уровень эстрогенов в крови. Представительницам женского пола прописывают Этинилэстрадиол, а мужского – Метилтестостерон.
- Гемокомпонентное лечение – переливание крови, тромбоцитарной массы.
- Лекарства железа (Ферромед, Актиферрин, Феррум Лек, Ферроплекс).
Зачастую лечение включает оперативное вмешательство. В процессе операции врачи удаляют пораженный участок сосуда, или ставят вместо него протез.
Что касается народных средств лечения, они прекрасно дополняют основное. В них входят полезные настои и травы, которые стимулируют метаболизм, повышают кровяную свертываемость и улучшают регенеративную функцию. Самые эффективные растения – крапива, тысячелетник, земляника и спорыш.
Профилактика
Основные меры профилактики болезни Рандю-Ослера:
- Консультация врача семей, которые планируют беременности;
- Закаливание организма;
- Соблюдение диеты;
- Постоянный контроль пульса и АД;
- Устраивать частые прогулки пешком;
- Полный отказ от вредных привычек;
Прогноз у больных при синдроме Рандю-Ослера в принципе благоприятный, однако существуют и такие формы, при которых очень непросто контролировать кровотечения.
- К каким докторам следует обращаться если у Вас Болезнь Рандю - Ослера
Что такое Болезнь Рандю - Ослера
Болезнь Рандю - Ослера - наиболее частая наследственная геморрагическая вазопатия с очаговым истончением стенок и расширением просвета микрососудов, неполноценным местным гемостазом. Наследуется данная патология по аутосомно-доминантному типу с различной встречаемостью патологического гена.
Что провоцирует Болезнь Рандю - Ослера
Причина заболевания до сих пор остается неизвестной. Что касается механизма развития, то по современным представлениям болезнь Рандю - Ослера - пример сосудистой патологии (с расстройством анатомической целостности сосудов) с врожденной тенденцией к недостаточному развитию кровеносных сосудов (сосудистая дисплазия). Такая аномалия характеризуется неполноценностью мезенхимы. Анатомическим субстратом болезни является истончение сосудистой стенки с отсутствием эластической и мышечной оболочек. Поэтому стенка представляет собой один эндотелий и окружена рыхлой соединительной тканью. В результате этого появляются артериовенозные аневризмы, которые из-за легкой ранимости сосудистых стенок дают кровотечения.
Чаще всего болезнь морфологически обусловлена наличием множественных телеангиэктазий, локализующихся на коже, слизистой языка, носа, бронхов, желудочно-кишечного тракта. Во многих случаях болезнь Рандю - Ослера носит наследственный характер, передается по доминантному типу. Встречаются однако и спорадические случаи заболевания.
Симптомы Болезни Рандо - Ослера
Хотя неполноценность мезенхимальной основы микрососудов генетически обусловлена, телеангиэктазий в раннем детском возрасте не видны и начинают формироваться лишь к 6-10 годам, чаще всего на крыльях носа, слизистой оболочке губ, десен, языка, щек, коже волосистой части головы и ушных мочек. С возрастом число и распространенность ангиэктазий возрастают, кровоточивость из них встречается чаще и бывает тяжелее.
В классическом описании В. Ослера выделены три типа телеангиэктазий:
- ранний - телеангиэктазий в виде небольших неправильной формы пятнышек;
- промежуточный - в виде сосудистых паучков;
- узловатый тип - в виде ярко-красных круглых или овальных узелков диаметром 5-7 мм, выступающих над поверхностью кожи или слизистой оболочки на 1-3 см.
У больных старше 25 лет часто обнаруживаются ангиэктазии 2 или всех 3 типов. Все они отличаются от других образований тем, что бледнеют при надавливании и наполняются кровью после прекращения давления.
У большинства больных телеангиэктазии сначала появляются на губах, крыльях носа, на щеках, над бровями, на языке, деснах, слизистой оболочке носа (при риноскопии плохо выявляются даже при кровотечениях). Затем они могут обнаруживаться на любых участках кожи, включая волосистую часть головы и кончики пальцев. Иногда они хорошо видны под ногтями, могут образовываться и на слизистых оболочках зева, гортани, бронхов, на всем протяжении желудочно-кишечного тракта, в почечных лоханках и в мочевыводящих путях, во влагалище.
В большинстве случаев геморрагические явления начинаются с носовых кровотечений, очень склонных к обострениям. Может долго кровоточить лишь один носовой ход, а иногда чередуются кровотечения разных локализаций.
Интенсивность и длительность кровотечений очень различны - от сравнительно необильных и не очень продолжительных до чрезвычайно упорных, длящихся почти беспрерывно в течение многих дней и недель, приводящих к крайней анемизации больных. Известны случаи, когда, несмотря на квалифицированную оториноларингологическую помощь, больные умирали от носовых кровотечений.
Такие же упорные и опасные кровотечения наблюдаются и из телеангиэктазии другой локализации: легочно-бронхиальной, желудочно-кишечной. Диагноз в подобных случаях устанавливается при эндоскопическом исследовании. В отдельных случаях зарегистрированы также кровоизлияния в мозг и во внутренние органы.
Врожденная неполноценность сосудов внутренних органов проявляется артериовенозными аневризмами, которые чаще всего локализуются в легких. При этом клиника включает в себя появление одышки, полиглобулии, у больных отмечаются цианотично-красный цвет лица, инъекция сосудов склер. Реже аневризмы появляются в печени, почках, селезенке. Эти артериовенозные аневризмы с трудом распознаются, часто трактуются как другие заболевания (от эритремии до туберкулеза или опухолей легких, врожденных пороков сердца). Продолжительно существующий ангиоматоз органов может привести к тяжелым и необратимым изменениям в них прогрессированию легочно-сердечной недостаточности, хронической почечной недостаточности. Но среди причин смерти особенно преобладают упорные кровотечения с тяжелой постгеморрагической анемией и сердечной недостаточностью.
Диагностика Болезни Рандо - Ослера
Диагностика болезни Рандю - Ослера не представляет трудностей при видимых телеангиэктазиях. Диагностике способствуют эндоскопические исследования.
Исследование системы гемостаза не выявляет существенных нарушений, способных объяснить кровотечения. Возможны лишь вторичные реактивные изменения, обусловленные кровопотерей (умеренная гиперкоагуляция, тромбоцитоз), анемией или, наоборот, полиглобулией при артериовенозном шунте. Вместе с тем при множественных телеангиэктазиях возможно появление признаков внутрисосудистого свертывания крови (коагулопатия потребления) с тромбоцитопенией.
Лечение Болезни Рандо - Ослера
Появлению и усилению кровоточивости, особенно носовых кровотечений, способствуют риниты и другие воспалительные заболевания слизистых оболочек, на которых расположены телеангиэктазии, их механические травмы (даже весьма легкие), стрессовые ситуации, умственное и физическое перенапряжение, прием алкоголя и острой пищи, особенно с уксусом, который нарушает агрегацию тромбоцитов, прием ацетилсалициловой кислоты и других дезагрегантов (препаратов, препятствующих свертыванию крови), недостаточный сон, работа в ночное время. Все это нужно учитывать при воспитании детей и подростков с телеангиэктазией, выборе спортивных занятий, профессии, при трудоустройстве и др.
Для остановки кровотечений используют локальные воздействия. Тугие тампонады носа малоэффективны, травмируют слизистую оболочку, способствуют более обильным и опасным последующим кровотечениям, возникающим тотчас или вскоре после удаления тампона. Местные орошения кровоточащей слизистой оболочки тромбопластином, тромбином, лебетоксом (стипвен, рептилаза), перекисью водорода недостаточно надежны и в лучшем случае лишь на некоторое время останавливают или уменьшают кровотечение. Лучший эффект дает орошение полости носа (с помощью резиновой груши или шприца) охлажденным 5-8%-ным раствором аминокапроновой кислоты, который всегда должен быть у больного в холодильнике.
Прижигания слизистой оболочки носа (трихлоруксусной и хромовой кислотами, нитратом серебра, диатермокоагуляцией) не предупреждают повторных кровотечений, а в ряде случаев способствуют их учащению. Временный эффект дают отслойка слизистой оболочки носа и перевязка приводящих артерий - верхнечелюстной, решетчатой. Тем не менее к таким вмешательствам приходится прибегать по жизненным показаниям при профузных кровотечениях. Из локальных воздействий более эффективно замораживание.
Первичную остановку кровотечения обеспечивает введение в полость носа гемостатического тампона или сжатой поролоновой губки, смоченной в жидком азоте. На втором этапе производят деструкцию телеангиэктазии с помощью криоаппликатора с циркуляцией азота (температура наконечника -196°С); время каждого замораживания -30-90 с. На третьем этапе проводят 4-8 сеансов (с промежутками в 1-2 дня) односекундных распылений жидкого азота в полости носа, что устраняет сухость слизистой оболочки и образование на ней корок. Продолжительность эффекта такого лечения колеблется от нескольких месяцев до 1 года и более.
К хирургическому лечению приходится прибегать при частых и очень обильных желудочно-кишечных, бронхо-легочных, почечных и других кровотечениях. Однако в связи с формированием новых ангиэктазий через некоторое время эти кровотечения могут возобновляться.
Общие терапевтические воздействия при болезни Рандю - Ослера малополезны. Геморрагический синдром иногда смягчается при назначении эстрогенов или тестостерона (гормональных препаратов). Викасол, хлорид кальция, желатин, гемофобин, дицинон, аминокапроновая кислота не купируют и не предотвращают кровотечений.
Артериовенозные аневризмы следует удалять хирургическим путем возможно раньше, до развития необратимых циркуляторных и дистрофических изменений в органах.
Болезнь Рандю-Ослера-Вебера, клинический случай.
Болезнь Рандю-Ослера-Вебера - это заболевание с клинической картиной геморрагического диатеза. Синоним «наследственная геморрагическая телеангиэктазия» (НТГ) в полной мере отражает сущность заболевания, для которого наиболее характерной считается триада признаков: телеангиэктазии на коже, слизистых оболочках и во внутренних органах; склонность к кровотечениям; аутосомно-доминантный тип наследования . Заболевание манифестирует с телеангиэктазий на коже и артериовенозных мальформаций. Они могут поражать носоглотку, ЦНС, легкие, печень, селезенку, мочевыделительную систему и желудочно-кишечный тракт.
Историческая справка
В 1864 году Саттон описал заболевание, проявляющееся носовыми кровотечениями и поражением сосудистой системы, которое в дальнейшем получило названия болезнь Рандю-Ослера-Вебера и наследственная геморрагическая телеангиэктазия. Английский врач Бенджамин Гай Бабингтон заметил наследственный характер данного заболевания в своей статье «Наследственное носовое кровотечение» в 1865 году.
Французский врач Рандю первым подчеркнул бледность кожных покровов и множественные ангиомы у больных с наследственной геморрагической телеангиэктазией и дифференцировал эту болезнь с гемофилией. и Фредерик Вебер опубликовали подробное описание данного синдрома, который впоследствии получил их имена.
С развитием гастроинтестинальной эндоскопии Реншоу описал «яркие красные просяные пятна размером с булавочную иголку» на слизистой, характерные для наследственной геморрагической телеангиэктазии.
Генетические аспекты
Болезнь Рандю-Ослера-Вебера отличается аутосомно-доминантным типом наследования. Если один из родителей является носителем патологии, то теоретически каждый ребенок в семье имеет 50% вероятности приобрести патологический ген. Согласно родословным, представленный тип наследования подтвержден в 87% случаев. В тех редчайших случаях, когда оба родителя страдают рассматриваемой патологией, можно с полной уверенностью говорить о 100% вероятности иметь в поколении ребенка с очень тяжелым течением заболевания.
К настоящему времени описаны четыре генетические формы данного заболевания. При первом варианте (НГТ1) имеется мутация гена эндоглина. Отмечено, что этот ген располагается на длинном плече хромосомы 9 (9q33-q34,1). Второй вариант (НГТ2) вызван мутацией гена АЛК1 (activin-receptorlike kinase I), который располагается на хромосоме 12 вблизи от центромеры (12q11-q19). При обоих типах НГТ мутация приводит к болезни путем гаптотипической недостаточности. Как эндоглин, так и AЛK1 представляет собой клеточный поверхностный рецептор, участвующий в сигнальных механизмах, опосредованных трансформируемым фактором роста. Мутации генов эндоглина и AЛК1 приводят к снижению концентрации рецепторов на поверхности эндотелия сосудов.
Недавно доказано существование третьего варианта (НГТ3), связанного с геном, расположенным на хромосоме 5. Четвертый вариант – редкая форма, сходная с ювенильным полипозом, видимо, вызывается мутацией в гене SHAD4.
Патогенез
В патогенезе заболевания ведущую роль играют анатомические и функциональные изменения сосудов. Анатомическая сущность болезни заключается в поражении кровеносных сосудов различного калибра с дегенерацией и гипоплазией мышечного и эластического слоев. В результате развивается очаговое истончение стенок, затем расширение просвета микрососудов. Кровоточивость объясняется чрезвычайно легкой ранимостью сосудистой стенки в месте ангиэктазии, а также повышением фибринолитической активности крови.
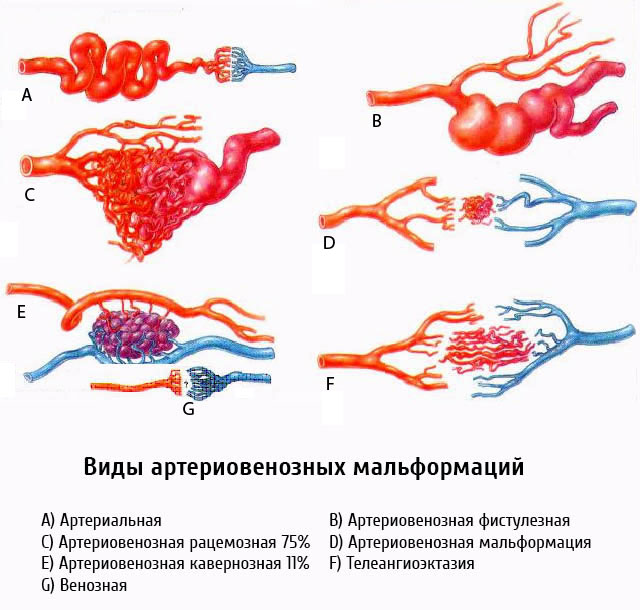
Сосудистые мальформации при НГТ (рис.1) состоят из прямых артериовенозных соединений через тонкостенные аневризмы и варьируют от небольших телеангиэктазий до огромных АВМ, располагающихся во внутренних органах.
На ранней стадии патологического процесса развивается локальная дилатация, вовлекающая все слои стенки посткапиллярной венулы. Затем это растяжение распространяется на артериолу, что в итоге приводит к формированию прямого их соединения без образования промежуточной капиллярной сети. Результатом этого процесса является снижение давления крови и скорости ее тока. При болезни Рандю–Ослера–Вебера наблюдается ангиоматозный тип кровоточивости с вторичным нарушением адгезивно-агрегационных функций тромбоцитов.
Нельзя исключить роль активации внутрисосудистого фибринолиза в зоне телеангиэктазий. В тяжелых случаях при заболевании отмечены признаки болезни фон Виллебранда, синдрома диссеминированного внутрисосудистого свертывания (ДВС-синдрома).
https://pandia.ru/text/78/059/images/image002_108.gif" width="183" height="230 src=">Вовлечение" href="/text/category/vovlechenie/" rel="bookmark">вовлечение дыхательной системы и печени (артериовенозные мальформации) (30%);
4. желудочно-кишечные кровотечения (15%) ;
5. поражения ЦНС .
Известны случаи дебюта заболевания с приступом кровавой рвоты, повторной макрогематурии. В литературе имеется единственное сообщение о начале болезни Рандю - Ослера - Вебера плачем «кровавыми слезами».
Однажды начавшись, кровотечения склонны к рецидивированию, причем интенсивность и длительность кровотечений индивидуальны (однократная кровопотеря составляет от нескольких капель до 500 мл и более). Переливание крови необходимо 10-30% больных.
Телеангиоэтазии обычно появляются через год после первого носового кровотечения.
По частоте развития кровотечений из внутренних органов при НТГ на первом месте стоят желудочно-кишечные кровотечения, затем легочные и почечные. Кровоизлияния в головной и спинной мозг наблюдаются у 2–3 % больных. Риск желудочно-кишечных кровотечений увеличивается у больных после 50 лет. Безболезненные кровотечения из гастроинтестинального тракта наблюдаются у 10-40% пациентов. Боль в животе может сопровождать тромбозы артериовенозных мальформаций в ЖКТ.
Легочные артериовенозные мальформации клинически могут проявляться цианозом, гипоксемией, одышкой, усиливающейся при физической нагрузке, могут наблюдаться кашель, кровохарканье, боли в грудной клетке, вторичная полицитемией. Возможно отсутствие симптомов. При исследовании больного с АВАЛ при аускультации сердца обычно не выявляется патологии, а электрокардиографические данные указывают на блокаду правой ножки пучка Гиса и реже – на перегрузку правых отделов сердца. В некоторых случаях над зоной сосудистой аномалии можно прослушать систолодиастолический шум, постоянный или непостоянный, интенсивность которого меняется во время дыхательных движений. Рентгенография легких обычно является начальным ведущим методом диагностики АВАЛ, однако отмечено, что не всегда, особенно при микроангиошунтировании, этот метод позволяет выявить сосудистую аномалию. Чаще всего рентгенологически регистрируются одиночные пульсирующие тени в легочных полях округлой, овальной или неправильной формы с четкими контурами. Для диагностики АВМ применяют также контрастную эхокардиографию.
Наиболее информативным методом обследования служит ангиопульмонография (АПГ), которая должна осуществляться при подозрении на множественные аневризмы, когда они имеют мелкофокусный вид образований небольших размеров от нескольких миллиметров до 1–2 см в диаметре.
По мере прогрессирования заболевания вследствие повторных кровотечений развивается хроническая железодефицитная анемия . В отдельных случаях формируются четкие клинические проявления дефицита железа в организме (сухость кожи, ломкость ногтей, волос).
В ответ на повторные геморрагии и развитие анемии можно отметить изменения сердечной деятельности. Имеются электрокардиографические признаки миокардиодистрофии. Эхокардиография у каждого пятого больного выявляет картину пролапса створок митрального клапана, что, видимо, отражает суть мезенхимальной патологии, характерной для данного заболевания; имеются признаки снижения показателей сократительной функции миокарда. При длительной постгеморрагической анемии развиваются признаки сердечной недостаточности, отечный синдром.
Диагноз НГТ ставится клинически на основании критерий Кюрасао, установленных в июне 1999 года. Они включают:
Носовые кровотечения (спонтанные, повторные);
Телеангиэктазии (множественные);
Висцеральные поражения (желудочно-кишечные с кровотечениями и без них), легочные артериовенозные аневризмы (ABA), спинальные ABA, сосудистые аномалии печени;
Аутосомно-доминантный тип;
Семейный характер.
При этом диагноз наследственной геморрагической телеангиэктазии является достоверным, если имеется, по крайней мере, три критерия; вероятным или возможным, если имеется два критерия; сомнительным, если имеется один критерий (Curacao, 1999).
Дифференциальная диагностика
Дифференциальная диагностика настоящего заболевания в общей клинической практике осуществляется с болезнями, сопровождающимися кровотечениями наследственного генеза (тромбоцитопатиями, болезнью Виллебранда и др.), а также с болезнями, при которых отмечаются телеангиэктазии (цирроз печени (таблица 1) и др.). Среди больных с носовыми кровотечениями НГТ имеет малый удельный вес - 0,5%, а среди острых желудочно-кишечных геморрагий – 0,1%. При висцеральном расположении сосудистых аномалий (в печени, легких) дифференциальный диагноз проводится с опухолями и туберкулезом.
Таблица 1. Дифференциальная диагностика с циррозом печени |
||
Болезнь Рандю-Ослера | Цирроз печени |
|
Наследственная природа заболевания | ||
Геморрагии из телеангиэктазий | ||
Форма телеангиэктазий | Часто точечные | Часто паукообразные |
Цвет телеангиэктазий | Красно-пурпурный | Ярко-красный |
Размер телеангиэктазий | До нескольких см в диаметре |
|
Количество телеангиэктазий | Десятки, сотни | |
Локализация телеангиэктазий | По всей поверхности туловища, на слизистых оболочках | На лице, грудной клетке, редко – ниже диафрагмы |
Сочетания с АВАЛ | ||
Функциональные нарушения печени |
Лечение
Одна третья случаев заболевания протекает в легкой форме, она третья – средней степени тяжести и одна третья – в тяжелой форме. При легкой форме лечение не требуется. Патогенетическая терапия болезни не разработана. В периоды кровотечений используют средства местной и общей гемостатической терапии - орошения тромбином с 5% раствором аминокапроновой кислоты, тампонада полости носа масляными тампонами, отслойка, слизистой оболочки в области кровотечения, прижигания. Более эффективна криотерапия. Иногда прибегают к хирургическому лечению (иссечение ангиом, пластика перегородки носа, перевязка и эмболизация артерий). Используют баротерапию, прижигания с помощью лазера. Все эти мероприятия часто дают лишь временный эффект. Введения викасола не показаны. При сопутствующем дефиците фактора Виллебранда показаны трансфузии свежезамороженной плазмы, введения криопреципитата. При анемизации - гемотрансфузии, введение препаратов железа.
Прогноз в большинстве случаев относительно благоприятный, но встречаются формы с неконтролируемыми кровотечениями.
Профилактика . Избегать травматизации слизистых оболочек в местах расположения ангиом, смазывание слизистой оболочки носа ланолином (с тромбином) или нейтральными маслами.
Клинический случай
Больной С., 83 лет был госпитализирован в 26 отделение ГКБ №4 с жалобами на слабость, носовое кровотечение. Ухудшение с 4.10.2011, когда возникло носовое кровотечение, не прекращающееся до 6.10.11. Утром 6.10.2011 отмечал рвоту типа «кофейной гущи». Бригадой СМП был доставлен в ГКБ №4 с диагнозом носовое кровотечение, ЖКК? Был осмотрен хирургом - данных за острую хирургическую патологию не было.
Анамнез заболевания. В 1972 году впервые установлен диагноз болезни Рандю-Ослера-Вебера, по поводу которого пациент в течение длительного времени наблюдался гематологами больницы им. Боткина. В клинической картине преобладают частые рецидивирующие носовые кровотечения, по поводу которых требуется переливание эритроцитарной массы и свежезамороженной плазмы. В течение последних 15 лет выявляется эрозивно-геморрагический гастрит. В течение 2009 года 6 госпитализаций с явлениями носовых кровотечений. Амбулаторно принимал препараты железы, на фоне которых появились боли в эпигастрии. Первая госпитализация в ГКБ №4 20.12.2009, в связи с рецидивирующим носовым кровотечением, рвотой «вишневой » кровью. Был госпитализирован в ОРИТ, по данным ЭГДС - диффузный эрозивный гастрит, бульбит. Семейный анамнез . Дочь и внучка больного страдают обильными частыми маточными кровотечениями.
Локальный статус. При осмотре слизистой языка (рис.2), на кожных покровах дистальных фалангах пальцев рук определяются множественные телеангиэктазии. (рис. 3).


Сопутствующие состояния :
· в 1952 г.-холецистэктомия по поводу гангренозного холецистита, после чего пациенту был установлен диагноз (со слов) постхолецистэктомического синдрома.
· В течение последних 20 лет повышения АД до 200/100 мм рт. ст. С того же времени перебои в работе сердца. При обследовании в сентябре 2009 года выявлена пароксизмальная форма мерцательной аритмии на фоне атеросклеротического стеноза АК. На данный момент – мерцательная аритмия, постоянная форма.
· Длительное время явления сердечной недостаточности. Принимает дигоксин, фуросемид, верошпирон.
· При обследовании в сентябре 2009 года при УЗИ щитовидной железы выявлено: в правой доле гетерогенный узел 36х54 мм с ровными контурами и гипоэхогенным ободком, в левой доле гипоэхогенные узлы 12 и 13 мм в диаметре.
Соматический статус . Пониженного питания. В легких жесткое дыхание, влажные мелкопузырчатые хрипы с обеих сторон, ЧДД - 18 в мин. Тоны сердца звучные, аритмичные систолический шум во всех точках аускультации. ЧСС - 80 в мин, АД - 120/60 мм рт. ст. Живот мягкий, безболезненный; печень и селезенка не увеличены. Стул и мочеиспускание не нарушены. Отеки до средней трети голеней и стоп.
Дополнительные исследования . В клиническом анализе крови обращало на себя внимание снижение Hb до 47 г/л, эритроцитов 2,9х1012/л. По рентгену органов грудной клетки – левосторонняя нижнедолевая пневмония, левосторонний гидроторакс, признаки легочной гипертензии.
По УЗИ брюшной полости, почек – диффузные изменения печени, поджелудочной железы, расширение воротной и селезеночной вен. Кисты почек. На ЭГДС – диффузный эрозивный гастрит, бульбит.
При нахождении в больнице пациент упал в связи с головокружением, на рентгене костей – чрезвертельный перелом левой бедренной кости со смещением.
По данным Эхо-КГ сердца: Гипертрофия миокарда левого желудочка. Митральная и трикуспидальная недостаточность 2ст., аортальный стеноз 1ст. Легочная гипертензия 2ст.
Коагулограмма при поступлении: АЧТВ 59,9 МНО 1,84 ПТИ 61,8%
Лечение:
Произведено переливание 4 порций эритроцитарной массы и 4 порций свежезамороженной плазмы. На фоне проведенных переливаний отмечалось улучшение состояния больного, подъем Hb от 47 г/л при поступлении до 75 г/л.
Также проводилось лечение сопутствующих состояний:
· Носовые кровотечения – гемостатическая терапия, передняя тампонада носа;
· Сердечная недостаточность – мочегонная, гипотензивная, противоаритмическая терапия.
· Пневмония – антибактериальная терапия.
Обсуждение. Учитывая жалобы пациента, анамнез, данные осмотра, данные лабораторных методов обследования у нашего пациента имели место:
Болезнь Рандю-Ослера-Вебера – достоверно доказанная, согласно критериям Кюрасао, с наличием носовых кровотечений, телеангиэктазий, семейного характера.
Осложнения: Хроническая анемия смешанного генеза (постгеморрагическая и железодефицитная), тяжелой степени тяжести.
Сопутствующие состояния: Гипертоническая болезнь 2 ст, риск 4. Комбинированный атеросклеротический митрально-аортальный порок сердца: стеноз и недостаточность. Постоянная форма мерцательной аритмии. Левосторонний гидроторакс. НК 2Б. Внегоспитальная левосторонняя нижнедолевая пневмония. Закрытый чрезвертельный перелом левой бедренной кости со смещением. Хронический эрозивный гастрит, бульбит, ремиссия. Многоузловой зоб, эутиреоз. Состояние после давней холецистэктомии. Фиброз печени. Портальная гипертензия. Кисты почек.
Обращает на себя внимание наличие большого количества тяжелых патологий у нашего пациента и преклонный возраст. Из анамнеза следует, что больной живет с доставерным диагнозом наследственной геморрагической телеангиоэктазии уже 39 лет. В связи с этим, можно сделать вывод о благоприятном течении заболевания при наличии адекватной симптоматической терапии.
Список литературы
1. , О. Л.И. и.с., 2002.
2. Guttmacher AE, M. D., White RI Jr. . Hereditary hemorrhagic telangiectasia. N Engl J Med., Oct 5 1995. 333 (14): p. 918-924.
3. HG., S., Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Med Mirror. , 1864: p. 769-81
4. BG., B., Hereditary epistaxis. . Lancet. , 1865: p. 362-363.
5. M., R., Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muquex. . Bull Mem Soc Med Hop Paris, 1896. 13 : p. 731.
6. W., O., On a family form of recurring epistaxis associated with multiple telangiectases of the skin and mucous membranes.
. Bull Johns Hopkins Hosp., 1901. 12 : p. 333-337.
7. FP., W., Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages. Lancet., 1907. ii : p. 160-162.
8. JA., S., Angiomatous conditions of the gastro-intestinal tract. Br J Surg. Mar 1953. 40 (163): p. 409-21.
9. Grover S, G. R., Verma R, Sahni H, Muralidhar R, Sinha P. , Osler-Weber-Rendu syndrome: a case report with familial clustering. Indian J Dermatol Venereol Leprol., Jan-Feb 2009. 75 (1): p. 100-101.
10. , Т. Н.В., Наследственнная геморрагическая телеангиоэктазия (болезнь Рандю -Ослера -Вебера ). Болезни сердца и сосудов, 2009. 4 (3): p. 65-70.
11. Braverman IM, K. A., Jacobson BS. , Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. . J Invest Dermatol., Oct 1990. 95 (4): p. 422-427.
12. В.Б. Золотаревский и соавт., Г. Д.Б. и.с., 2001.
13. Begbie ME, W. G., Shovlin CL. , Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J., Jan 2003. 79 (927): p. 18-24.
14. Fulbright RK, C. J., Putman CM, et al. , MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. . AJNR Am J Neuroradiol., Mar 1998. 19 (3): p. 477-484.
15. Jakobi P, W. Z., Best L, Itskovitz-Eldor J. , Hereditary hemorrhagic telangiectasia with pulmonary arteriovenous malformations. . Obstet Gynecol., May 2001. 97 (5 Pt 2): p. 813-814.
16. Shovlin CL, G. A., Buscarini E, et al., Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet, Mar 6 2000. 91 (1): p. 66-67.